Sjúkurnar
Í Føroyum finnast fleiri lívshættisligar ílegusjúkur. Samanborið við onnur lond, koma hesar sjúkur óvanliga ofta fyri í Føroyum. Á hesi síðu lýsa vit tær mest hættisligu ílegusjúkurnar. Niðast á síðuni, undir evninum Arvamynstur, greiða vit frá, hvussu børn kunnu arva hesar frá foreldrunum.
Yvirlit
Felagið Arvasjúkan mælir til at seta á stovn eitt alment berarakanningartilboð fyri tær fimm ílegusjúkurnar, sum ikki kunnu viðgerðast. Hesar sjúkurnar eru deyðiligar og ólekiligar, og einasta hjálp fyri børnini er linnandi viðgerð. Nøkur børn doyggja stutt aftaná føðing og nøkur fá liva inn í tannárini og eitt sindur longri. Felags fyri øll hesi børn og ungu er, at tey hava tørv á røkt og umsorgan alt døgnið.
Mett verður, at í hvørjum skúlaflokki eru um 5 næmingar, sum eru berarar av einari av teimum 5 sjúkunum uttan viðgerðarmøguleikar.
| Stytting | Navn | Miðal lívstíð | Beraratíttleiki |
|---|---|---|---|
| AGS | Aicardi-Goutières syndrome | 3,5 ár | 1 út av 22 |
| ARPKD | Autosomal recessive polycystic kidney disease (Potters syndrome) | Nakrar fáar dagar | 1 út av 25 |
| JEB | Junctional epidermolysis bullosa, generalized severe | Minni enn 1 ár | 1 út av 30 |
| SUCLA2 | Succinyl-CoA synthetase deficiency (Føroyska sjúkan) | Uml. 9 ár | 1 út av 23 |
| TBCD | Tubulin-binding cofactor deficiency | 26 mánaðir | 1 út av 30 |
Afturat teimum fimm ílegusjúkunum omanfyri, lýsa vit fýra aðrar deyðiligar ílegusjúkur, sum eisini koma serliga ofta fyri í føroyum. Hesar sjúkurnar kunnu tó viðgerðast og sjúklingarnir kunnu liva meira ella minni uttan árin av sjúkunum.
| Stytting | Navn | Miðal lívstíð | Beraratíttleiki |
|---|---|---|---|
| CF | Cystisk fibrose | 30 - 40 ár við viðgerð | 1 út av 17 |
| CTD | Carnitine transporter deficiency | Vanlig við viðgerð | 1 út av 9 |
| GSD | Glycogen storage disease | Vanlig við viðgerð | 1 út av 30 |
| HLCS | Holocarboxylase synthetase deficiency | Vanlig við viðgerð | 1 út av 23 |
AGS
Aicardi-Goutières syndrome
Sjúkan kann hvørki lekjast ella viðgerðast. Børnini eru likamliga og kognitivt avlamin - tey eru fullkomiliga óhjálpin og hava tørv á at verða ansaði alt døgnið. Viðgerðin er bert linnandi.
Sjúkueyðkennir
Sjúkueyðkennini vísa seg vanliga innan teir fyrstu 3 mánaðirnar aftaná barnið er borið í heim. Tey fyrstu sjúkueyðkennini plaga at koma til sjóndar við at børnini spýggja nógv, eru órólig og hava trupult við at eta. Seinni koma tekin sum krampaherðindir, spastiskar rørslur í armar og bein, ónormalar eygnarørslur og sein likamlig og kognitiv menning. Børnini hava lítið høvd orsaka av, at heilin ikki veksur sum hann eigur at gera. Hetta kann staðfestast vil einari MR skanning.
AGS er ein tann mest álvarsama ílegusjúkan, sum vit kenna til í Føroyum. Ílegubroytingar, sum føra við sær AGS, eru eyðmerktar aðrastaðni í heiminum, men føroyska frábrigdi vísir seg at vera ímillum tey meira álvarsomu, hvat sjúkueyðkennum viðvíkur.
Av tí at fosturdeyði, orsaka av AGS, er staðfestur í Føroyum, og tí at talið av berarum av AGS brekinum, í Føroyum, er stórt, er tað viðkomandi at hava í huga, um orsøkin til ein fosturdeyða ella pinkubarnadeyða, millum annað, kann stava frá hesi sjúkuni.
AGS kann líkjast viðføddum heilabruna. Í slíkum føri er tað viðkomandi eisini at kanna barnið fyri AGS.
Frágreiðing um sjúkuna
Føroyska fyribrigdi av AGS stavar frá einari broyting í RNASEH2B íleguni. RNASEH2B er uppskriftin til enzymið RNase H2, ið hevur til uppávu at rudda upp í ávísum feilum, sum kunnu koma í DNA aftaná at tað er kopierað. Tá enzymið ikki megnar at gera sína uppgávu til fulnar, vil immunverjan tulka kyknur við DNA feilum sum fremmandalikam og reagera við at royna at beina tær burtur. Hetta førir til eina autoimmuna støðu, har immunverjan loypur á egnan kropp. Avleiðingin er ein brunalíknandi støða í heilanum og øðrum organum, sum fáa stóran skaða av hesum.
ARPKD
Autosomal recessive polycystic kidney disease (Potters syndrome)
Sjúkan kann hvørki lekjast ella viðgerðast. Børnini kunnu ikki liva við hesari sjúku.
Sjúkueyðkennir
Ultraljóðskanningin av barnakonum, sum verður gjørd í 20. viku, kann vísa, um nýrini hjá fostrinum eru størri enn vanligt og hava í sær bløðrir (cystur), sum eru fullar av vatni. Ultraljóðskanningin kann eisini vísa nógva vætu í heilanum og øðrum organum. Hesi eyðkennir eru vanliga tekin til ARPKD sjúkuna.
Skanningin vísir eisini, at mongdin av fosturvatni er nógv minni enn vanligt, tí nýruni hjá fostrinum ikki megna at framleiða neyðugu vætuna til fosturvatnið. Lítla mongdin av forsturvatni gevur fostrinum ov lítið av plássi til at vaksa í. Hettar kann føra við sær, at andlit og aðrir likamslutir hjá barninum fáa eitt kroyst skap.
Børn, sum verða fødd við hesi sjúku, liva ikki leingi; bert ein dag ella tveir, tí at nýruni ikki megna at reinsa blóðið og/ella at lunguni ikki eru fullment. Fleiri børn doyggja í móðurlívið.
Frágreiðing um sjúkuna
PKHD1 ílegan er uppskriftin til proteinið fibrocystin, sum serliga er at finna á kyknuveggjum í nýravevnaðinum. Fibrocystin hevur ein lyklaleiklut í menningini av vevnaðinum í nýrunum. Broyting í hesi ílegu hevur við sær, at nýrini ikki útviklast sum tey eiga og persónurin fær sjúkuna Autosomal recessive polycystic kidney disease (ARPKD).
CF
Cystisk fibrose
Sjúkan kann ikki lekjast, men tað finnst heilivágur, sum kann linna sjúkueyðkennini.
Sjúkueyðkennir
Sjúkueyðkennini við CF eru tarmtrupulleikar hjá nýføddum, feittkent diarré, vantandi vøkstur og afturvendandi lungnabruni. Seinni kemur vanliga brisbruni, gallsteinur og diabetes. Lungakapasiteturin versnar við tíðini og tey flestu doyggja orsaka av vánaligari lungafunktión.
Viðgerðin er bæði tíðarkrevjandi og kostnaðarmikil. Føringar, sum hava CF, mugu ferðast til Danmarkar einaferð um mánaðan til kontrol. Við núverandi viðgerð er livialdurin umleið 30-40 ár.
Her skal leggjast afturat, at ein nýggj og munagóð viðgerð væntandi fer at føra við sær, at livialdurin hjá CF sjúklingum fer at hækka nógv.
Sigrun Jóhansdóttir Bisp hevur verið nógv frammi í miðlunum og greitt frá sínum lívi við CF. Hon fekk lungatransplantatión í februar 2017 og aftur í desembur 2019. Sigrun andaðist í februar 2023, 33 ára gomul.
Sigrun hevði eisini ein Facebook vanga, har hon regluliga legði út ymiskt um sítt lív við CF.
Frágreiðing um sjúkuna
Orsøkin til Cystiska fibrosu er, at eitt protein, ið situr í kyknuvegginum og skal hjálpa til við vætujavnvágini inn og úr kyknunum, ikki virkar sum tað skal. Tá verður ov nógv væta verður hildin aftur inni í kyknunum og ov lítið av vætu verður eftir úti ímillum kyknurnar. Hetta førir við sær, at ymisk væta í kroppinum gerst ov tjúkk. Tá vætan í lungunum og tarmunum, verður ov tjúkk fær persónurin niðursetta lunga og tarmfunktión. Eisini brisið (búkspýttkertilin) og livurin eru vanliga nógv ávirkað.
Cystisk fibrosa er ein vanlig sjúka í norðurevropa. Ein orsøk til hetta kann vera kolera sjúkan, sum herjaði í norðurevropa í 1800 talinum. Berarar av CF ílegubrekinum hava tjúkkari vætu í kroppinum enn onnur. Hetta kann vera ein orsøk til, at tey hava verið meira mótstøðufør ímóti kolera, tí kolera sjúkan førir við sær lívshóttandi vætutrot.
CTD
Carnitine transporter deficiency (karnitintrot)
Sjúkan kann ikki lekjast, men viðgerð við heilivági (karnitinískoyti) ger, at fólk kunnu liva uttan árin av sjúkuni.
Sjúkueyðkennir
Sjúkutekin, sum hjartaólag, kann vísa seg millum 1 og 7 ára aldur. Í ungdómsárunum vísir sjúkan seg ofta við álvarsligum rútmuólagi í hjartanum, vøddaveikleika ella vøddapínu. Eitt vanligt sjúkueyðkenni er máttloysi - serliga aftaná harða likamliga venjing ella aftaná at hava fasta í nógvar tímar.
Viðgerðin av CTD er støðug og reglulig inntøka av tablettum við karnitin 3-4 ferðir um dagin. Tá liva fólk mestsum óávirkaði av sjúkuni. Tað hevur sera stóran týdning, at fólk lata seg kanna fyri at finna útav, um tey mangla karnitin, tí viðgerðin er góð fyri hesa sjúku, og fólk, sum hava CTD og ikki eru í viðgerð við karnitinískoyti, kunnu doyggja brádliga av hjartasteðgi.
Eitt felag er sett á stovn við tí endamáli at upplýsa og leiðbeina um CTD. Á heimasíðu teirra kanst tú kunna teg meira um CTD.
Frágreiðing um sjúkuna
Í Føroyum finnast serliga nógvar hættisligar broytingar í SLC22A5 íleguni, ið kunna føra til sjúkuna Carnitine transporter deficiency (CTD).
Hesar ílegubroytingar gera, at eitt protein, ið skal hjálpa við at flyta karnitin inn í kyknurnar, ikki riggar sum tað skal. Tí verður trot á karnitini inni í kyknunum í kroppinum og karnitini verður ístaðin útskilt við urinini. Í kyknunum skal karnitinið nýtast til at flyta feittketur inn í mitokondriini (orkuverkið í kyknunum). Mitokondriini gera, at feittið verður niðurbrotið til tey evnir, sum kyknan skal brúka til orku. Tá ov lítið av karnitini er til staðar fær kyknan ov lítið av orku.
Serliga tá blóðsukrið er lágt er tað alneyðugt at karnitin er til staðar í kyknunum, tí tá noyðist kroppurin at brenna feitt fyri at fáa orku. Um kyknurnar í kroppinum ikki fáa nóg mikið av orku kann tað føra til, at heili, hjarta og vøddar, kunnu verða nógv ávirkaði og møguliga fáa skaða.
GSD
Glycogen storage disease (glukosugoymslubrek)
Sjúkan kann ikki lekjast, men serstakur kostur kann bøta um sjúkueyðkennini.
Sjúkueyðkennir
Sjúkueyðkennini vísa seg tíðliga í lívinum við at børnini hava lágt blóðsukur, óvanliga nógv feitt í blóðinum og hækkaða mongd av livurenzymum í blóðinum. Henda støða førir til, at børnini goyma nógv glycogen í kroppinum, sum ger at tey verða ovurfeit og fáa stóra livur og tískil stóran og rundan búk. Støddin á livrini kemur vanliga aftur á normala stødd í ungdómsárunum. Nakrir sjúklingar kunnu útvikla áhaldandi livrasjúkur og onnur livrabrek seinni í lívinum.
Fyrstu tekinini um sjúku eru ofta veikir vøddar í barndóminum. Vøddaveikleikin kann versna til ógvusligan veikleika tíðliga í vaksnamannaaldrinum ella á miðjum árum. Einstakir sjúklingar kunnu fáa veikar hjartavøddar (cardiomyopati), men vanliga førir sjúkan ikki til hjartabrek.
Sjúkan kan ikki lekjast, men fólk, sum hava GSDIIIA, kunnu fáa tað munandi betri, um tey hava eitt javnt blóðsukur, sum liggur innanfyri normaløkið; tað gerst við at eta fleiri ferðir um dagin – nógvar smáar máltíðir. Hesin matur skal, millum annað, verða settur saman við seint-upptakandi kolhydratum. Um sjúkan verður staðfest tíðliga er váðin fyri hjartasvik í barnaárunum munandi minni. Í summum førum kann livratransplantation gerast neyðug.
Frágreiðing um sjúkuna
Glycogen storage disease IIIa kemur av einari ílegubroyting, sum ger, at eitt enzym (glycogen debrancher enzyme GDE), sum skal klippa ávísar langar kolhydrat ketur (glycogen) sundur, ikki riggar sum tað skal. Glycogen verður nýtt til at goyma sukur (glucosu) í livrini, vøddum og hjartanum so tað kann nýtast til forbrenning seinni. Við GSDllla hópar glycogen seg upp og ger skaða á vevnaðin har tað verður goymt (livur, vøddar og hjarta). Tá glycogenið ikki kann klippast sundur til glukosu førir tað við sær, at ov lítið av sukri verður leysgivið út í blóðið og persónurin hevur tískil ov lítla orku.
HLCS
Holocarboxylase synthetase deficiency (biotintrot)
Sjúkan kann ikki lekjast, men heilivágur (biotinískoyti) ger, at fólk kunnu liva uttan árin av sjúkuni.
Sjúkueyðkennir
Sjúkueyðkennini koma vanliga til sjóndar teir fyrstu mánaðirnar av lívinum, men kunnu eisini vísa seg seinni. Børn við biotintroti hava ofta trupulleikar við at eta, eru trong, maktarleys, hava húðaskriða og missa hár. Óviðgjørd kann sjúkan føra til seinkaða menning og livshættisligar tilburðir við krampa og vitloysi.
HLCS kann viðgerðast við kostískoyti av biotin. Skjót og lívslong viðgerð kann fyribyrgja komplikatiónunum. Tí er tað umráðandi at kunna staðfesta biotintrot so skjótt sum til ber aftaná føðing. Berarar av HLCS ílegubroytingini hava ikki stórvegis árin.
Frágreiðing um sjúkuna
Broytingin í HLCS íleguni førir við sær, at eitt enzym, sum eitur holocarboxylase synthetase, bert riggar 20%. Hetta leiður til, at fólk fáa sjúkuna holocarboxylase synthetase deficiency. Enzymið hjálpur til at binda biotin (vitamin B7) til nøkur onnur enzym, ið hava brúk fyri biotin til at gera týðandi uppgávur í samband við niðurbrótan av proteini, feitti og kolhydratum. Biotin er vanliga at finna í livur, eggjablommu og mjólk.
Av tí at holocarboxylase synthetase enzymið riggar 20% í føroysku ílegubroytingini, so eru sjúkueyðkennini mildari enn sama sjúka aðrastaðni í heiminum, hvar holocarboxylase synthetase enzymið møguliga als ikki riggar.
JEB
Junctional epidermolysis bullosa, generalized severe
Sjúkan kann hvørki lekjast ella viðgerðast. Børnini hava bløðrur og sár í munninum, svølgrúminum, andingarleiðini og á húðini kring allan kroppinum. Hetta førir við sær sera vánaliga lívsgóðsku við nógvari pínu, trupulleika við at anda, álvarsomum sárum og ringari matarlyst. Viðgerðin er bert linnandi.
Sjúkueyðkennir
Børnini hava ikki nøkur serstøk sjónlig sjúkueyðkennir beinavegin tey eru fødd. Hetta kemst av, at barnið hevur verið vart í móðurlívi, tí fosturvatnið hevur forða fyri trýsti á húðina. So skjótt barnið er føtt kemur trýst á húðina - t.d. tá barnið liggur á einum undirlagið og skal latast í klæðir ella tá blæðan skal skiftast. Trýstið á húðina ger, at húðin dettur av og børnini fáa sár. Sárini lekjast, men húðin, sum kemur aftur, er framvegis leys. Hesi børn hava skiftandi sár ymsastaðni á kroppinum.
Børnini fáa eisini bløðrur/sár í munnin og í andingarleiðina, sum ger, at tey hava trupult við at eta og trívast tí illa. Tá sárini eru grødd kemur arrvevnaður í staðin. Hetta kann hava álvarslig árin á andingarleiðina soleiðis at børnini hava trupult við at anda og hava veika og hása rødd, tá tey gráta.
Við tíðini fáa børnini fleiri og størri opin sár, sum eru lívshóttandi. Sárini bløða illa og nógv væta rennur úr teimum, sum inniheldur salt og onnur lívsneyðug evnir. Bruni kann eisini lættliga koma í sárini. Børnini gerast so veik av hesum, at tey kunnu doyggja brádliga.
Í samband við eitt tiltak um forsturdiagnostik, sum Etiska Ráði skipaði fyri í 2019, segði Fróði Joensen, barnalækni á Landsjúkrahúsinum, soleiðis:
"Hettar er ein sjúka, sum er so álvarsom, at í okkara tankum, í læknaverðini, so er hettar ikki ein sjúka, sum børnini skuldu verið fødd við, um har var ein møguleiki fyri at stýra uttanum tað.”
“Hesi børnini hava vanliga eina livitíð á leið 6 mánaðir, har tey hava øgiliga pínu allatíð, sum svarar til at hava brandsár. Í hesari stuttu livitíðini hava tey onga lívsgóðsku, tá hugsa verður um, hvussu álvarsom og ring støðan hjá teimum er."
Frágreiðing um sjúkuna
JEB sjúkan kemur av einum ílegubreki, sum ger, at ein feilur er í proteininum laminin-332, sum hevur til uppgávu at festa húðina í vevnaðin niðriundir. Hettar merkir, at húðin liggur leys omaná og er sera viðbrekin - bert lætt trýst kann føra til bløðrur og at húðin dettur av. Hetta leiður til álvarsom og lívshóttandi sár, sum eru sera pínufull.
SUCLA2
Succinyl-CoA synthetase deficiency (Føroyska sjúkan)
Sjúkan kann hvørki lekjast ella viðgerðast. Børnini eru likamliga avlamin - tey eru fullkomiliga óhjálpin og hava tørv á at verða ansað alt døgnið. Nøkur børn eru órólig, hava pínu, gráta nógv, sveitta nógv v.m. Viðgerðin er bert linnandi.
Sjúkueyðkennir
Tey vanligu sjúkueyðkennini eru, at børnini hava nógv niðursetta hoyrn/eru deyv, kunnu ikki tosa, hava veikar vøddar og klára ikki at halda høvdinum sjálvi. Seinni mugu tey brúka korsett fyri at styðja ryggin. Tey klára ikki at ganga, tørva hjálp til at venda sær í songini, hava trupulleikar við andingarlagnum, vánaligan trivna, hava tørv á at sita í serligum stóli, sum stuðlar væl undir allan kroppin v.m. Tey flestu skulu hava flótandi føði ígjøgnum sondu, tí tey eru ov veik til at svølgja.
Børn, sum hava Føroysku sjúkuna, kunnu hava nógv laktat í blóðinum í eitt tíðarskeið. Henda støða kann føra við sær høvuðpínu, pínu í kroppinum, vaml, títtan andadrátt, nógvan sveitta og brádligan deyða.
Mentalt tykjast børnini ikki at vera ávirkað og eru tey tí tilvitað um, at tey eru sjúk. Fyri at bøta um lívsgóðskuna hjá børnunum kunnu tey, við skurðviðgerð, fáa ísett eitt Cochlear Implantat (hoyritól) so børnini kunnu hoyra. Tað finnast eisini onnur hjálpitól, sum kunnu hjálpa børnunum við at samskifta.
Børnini tola illa at verða sjúk og forkølað av vanligum smittusjúkum, sum fyri onnur er gerandiskostur. Tey kunnu fáa skaða á motoriksentrið í heilanum og gerast kropsliga verri fyri - í ringasta føri kunnu tey doyggja av hesum.
Hóast fyribyrgjandi tiltøk, so hevur hendan ógrøðandi sjúkan álvarslig árin á hjarta, lungur og onnur gøgn. Summi doyggja áðrenn tey eru eitt ár, men nøkur fá liva til tey eru í tjúgunum.
Frágreiðing um sjúkuna
SUCLA2 (Føroyska sjúkan) er ein ógvuliga sjáldsom stoffskiftisjúka, sum hevur høgan títtleika í Føroyum samanborið við onnur lond. Granskarar hava staðfest, at Føroyska sjúkan stavar frá einari ílegubroyting hjá einum persóni, sum livdi í Føroyum umleið ár 1630.
Føroyska sjúkan kemur av einari ílegubroyting, ið broytur skapið av einum enzymi soleiðis, at tað ikki riggar, sum tað skal. Enzymið skal nýtast í mitokondriunum (orkuverkinum) í kyknunum. Tá hetta ikki riggar fær mitokondrian bert framleitt umleið 20% av orkuni til kyknurnar. Tí fáa børnini ikki ta orku, sum tey hava tørv á fyri at liva og mennast væl.
Tá mitokondriini í kyknunum ikki fáa framleitt nóg nógva orku, brúkar kroppurin aðrar møguleikar til orkuframleiðsu. Tá kann tað henda, at mjólkasýra (laktat) eisini verður framleidd sum biprodukt. Hetta kann føra til, at barnið fær nógv laktat í blóðið. Laktat kenna tey flestu sum orsøkina til andsperri/pínu aftaná harða venjing.
TBCD
Tubulin-binding cofactor deficiency
Sjúkan kann hvørki lekjast ella viðgerðast. Børnini eru fjølbrekað - tey eru fullkomiliga óhjálpin og hava tørv á at verða ansað alt døgnið. Viðgerðin er bert linnandi.
Sjúkueyðkennir
Børn, sum verða fødd við TBCD troti, sýnast at vera frísk og mennast sum tey skulu fyrstu tíðina. Aftaná stutta tíð koma tekin um heilaskaða. Tey fáa veikar og linar vøddar, hava trupulleikar við at eta og svølgja og mennast ikki sum tey skulu. Seinni fáa børnini afturstig í menningini og missa førleikar, sum tey hava havt t.d. at sita, føra hendurnar upp til munnin og at halda høvdinum sjálvi. Tey fáa epileptisk herðindi, óstýrligar og, til tíðir, pínufullar vøddarørslur, niðursetta sjón ella gerast heilt blind. Frá at vera smábørn, sum pjátra og smílast til onnur, steðga tey við at vera varug við sítt umhvørvi.
Álvarsami og progressivi heilaskaðin førir við sær, at hesi børnini gerast likamliga fjølbrekaði og menningartarnaði. Børnini eru fullkomiliga óhjálpin og hava tørv á at verða ansaði alt døgnið. Sjúkan kann hvørki lekjast ella viðgerðast. Viðgerðin er bert linnandi. Hesi børn liva stutt, í miðal 26 mánaðir, og doyggja vanliga av lungabruna ella orsaka av vatni í lungunum, soleiðis at tey ikki fáa anda.
Frágreiðing um sjúkuna
Ein variantur av TBCD troti varð nýligani (í 2016) funnin í Føroyum. Hendan ílegubroytingin hevur verið orsøk til álvarsama og deyðiliga sjúku hjá minst 7 føroyskum børnum, sum eru fødd síðan 1991.
TBCD ílegan er uppskriftin til proteini Tubulin Folding Cofactor D, sum hjálpir til at skapa proteini tubulin. Tubulin verður sett saman til evarska smá rør kalla mikrotubuli, ið verða nýtt sum strukturur til nógv ymisk endamál inni í kyknunum hjá menniskjum. Hetta brekið í TBCD íleguni hevur við sær, at heilavevnaðurin minkar við tíðini. Heilabjálkin (hesin sambindur báðar heilahálvurnar) minkar eisini.
Arvamynstur
Víkjandi arvagongd
Menniskju hava tvey kopi av hvørjari ílegu (kyns-kromosonini eru undantikin). Tá eitt barn verður til í móðurlívið, arvar tað eitt kopi av øllum ílegunum frá hvørjum foreldri. Arvamynstrið í víkjandi arvagongd er soleiðis, at ávísar sjúkur bert koma til sjóndar, tá tvey kopi av sama ílegubrekinum eru til staðar.
Við øðrum orðum kunnu ein mamma og ein pápi bert fáa eitt sjúkt barn, tá barnið hevur arva tvey kopi av sama ílegubreki; eitt kopi frá hvørjum foreldri. Arva børnini bara eitt kopi av ílegubrekinum, gerast tey berarar av ílegubrekinum og eru sostatt ikki sjúk sjálvi. Myndlýsingarnar niðanfyri lýsa ymisk arvamynstir fyri børnini.
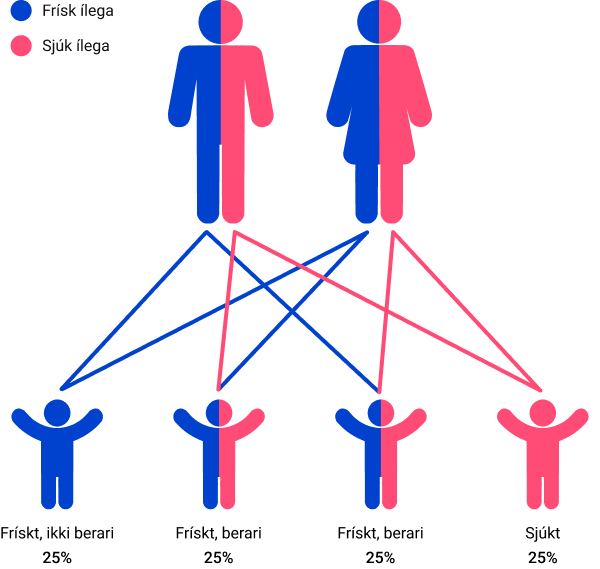
Bæði eru berarar
Sannlíkindini fyri hvørt barnið, tá bæði foreldrini eru berarar av sama ílegubreki:
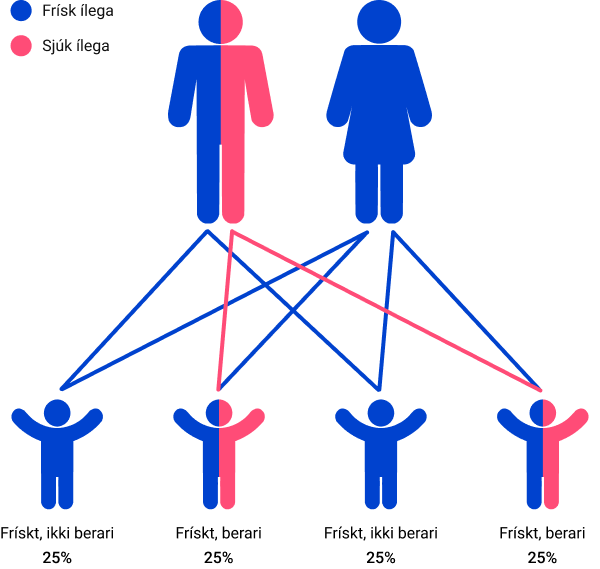
Annar er berari
Sannlíkindini fyri hvørt barnið, tá bara annað foreldrið er berari av einum ílegubreki:
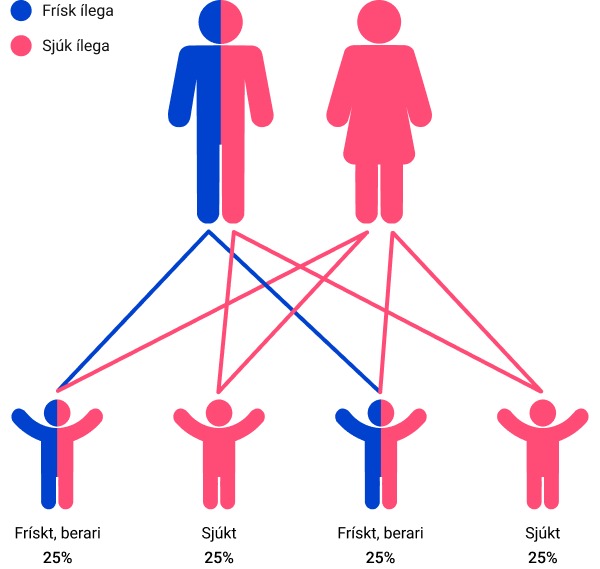
Ein ikki-berari og ein sjúkur
Sannlíkindini fyri hvørt barnið, tá annað foreldrið er ikki-berari av einum ílegubreki og hitt hevur sjúkuna:
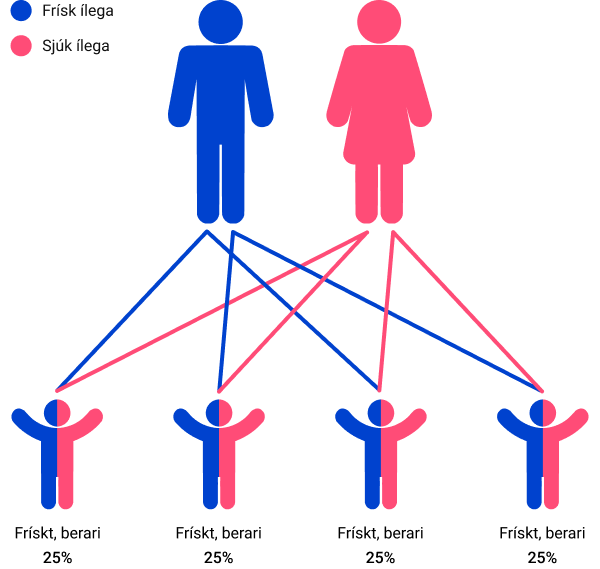
Ein berari og ein sjúkur
Sannlíkindini fyri hvørt barnið, tá annað foreldrið er berari av einum ílegubreki og hitt hevur sjúkuna: